
Qué tratamos
Nos sentimos honrados de servir a TODAS las personas con un trastorno hemorrágico
Atención integral para trastornos hemorrágicos
Hemofilia A
Hemofilia B
Portador de hemofilia
Enfermedad de von Willebrand
Deficiencias plaquetarias
Telangiectasia hemorrágica hereditaria
Trombastenia de Glanzmann
Otros trastornos hemorrágicos poco frecuentes
Hemofilia A y B
¿Qué es la hemofilia?
Existen dos tipos principales de hemofilia: la hemofilia A es una deficiencia del Factor 8 de coagulación y la hemofilia B es una deficiencia del Factor 9 de coagulación.
CUÁN COMÚN ES
La hemofilia afecta aproximadamente a 20.000 personas en Estados Unidos.
CAUSAS
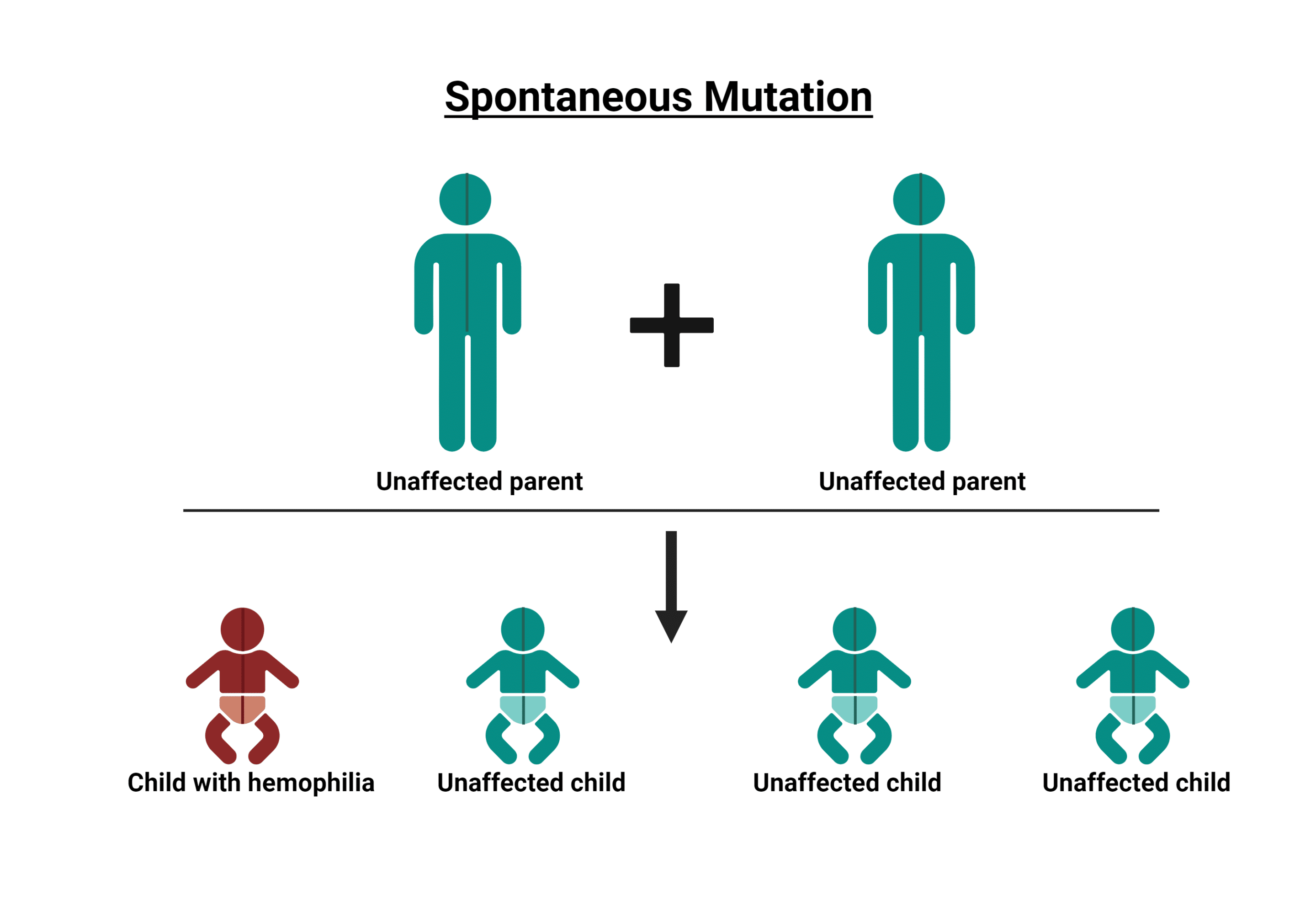
Este trastorno genético se caracteriza por la ausencia o deficiencia de una proteína de coagulación en el plasma, lo que provoca un retraso en la coagulación (hemorragia prolongada). La hemofilia se da casi exclusivamente en varones. Los hombres transmiten el gen defectuoso a sus hijas, que se convierten en portadoras de la hemofilia. Aunque existe una incidencia de mutación, el defecto genético se transporta en el cromosoma X. En consecuencia, las mujeres son portadoras de hemofilia y pueden padecer el trastorno y/o experimentar complicaciones hemorrágicas.
GRAVEDAD
La gravedad de la hemofilia A viene determinada por el porcentaje de Factor 9 que se encuentra en la sangre. Tenga en cuenta que la hemofilia es única en cada individuo, y que las hemorragias pueden no estar relacionadas con la gravedad (un hemofílico leve puede tener más hemorragias que uno grave).
- Leve – Entre el 5% y el 40%
- Moderado – Entre el 1% y el 5%
- Severe – <1%
SÍNTOMAS
Las personas con hemofilia corren el riesgo de sufrir hemorragias tras un traumatismo, una intervención quirúrgica o un tratamiento dental. También pueden tener hemorragias internas o articulares sin causa aparente. Cuanto mayor es la deficiencia del factor de coagulación, mayor es el riesgo de hemorragia.

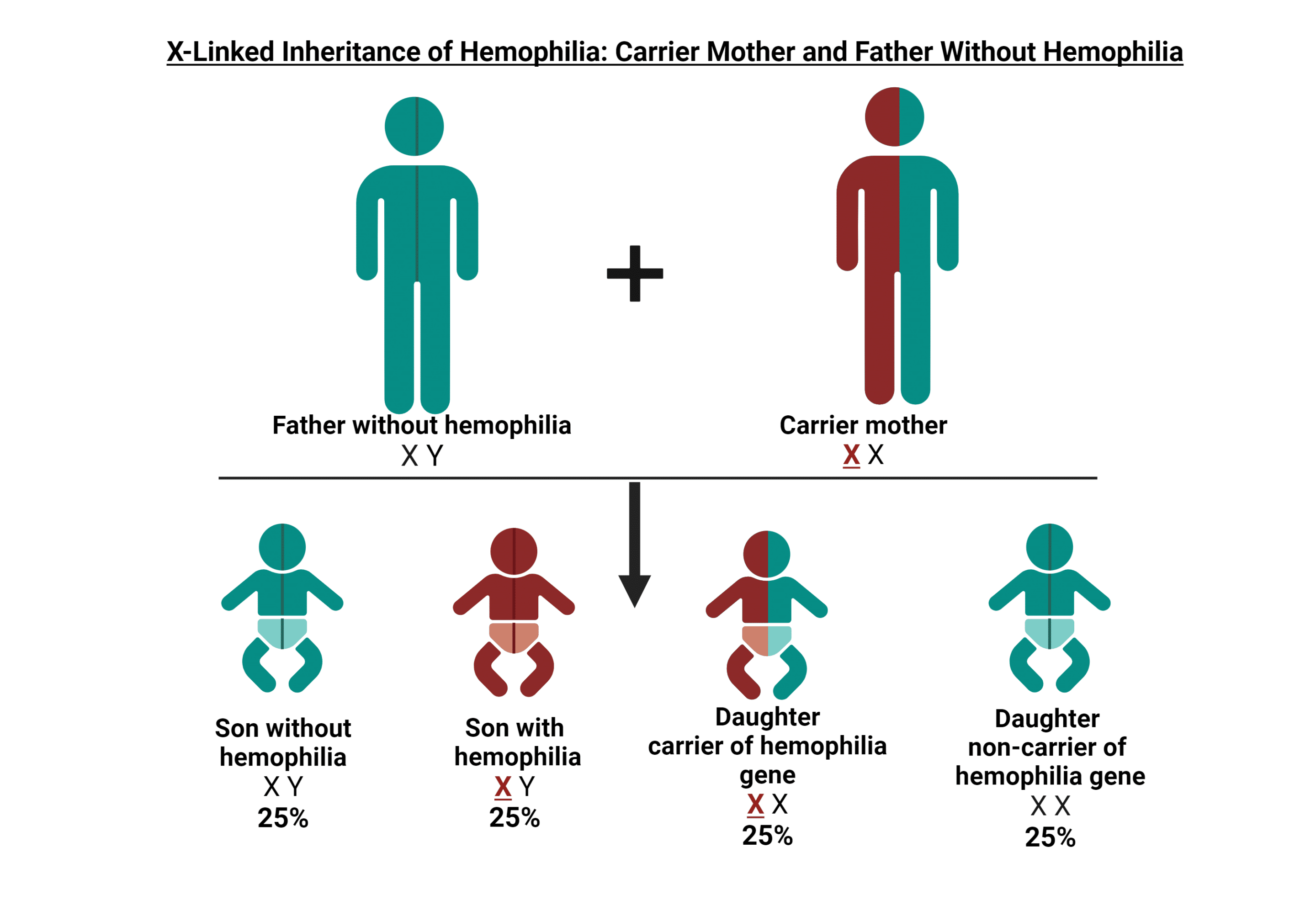
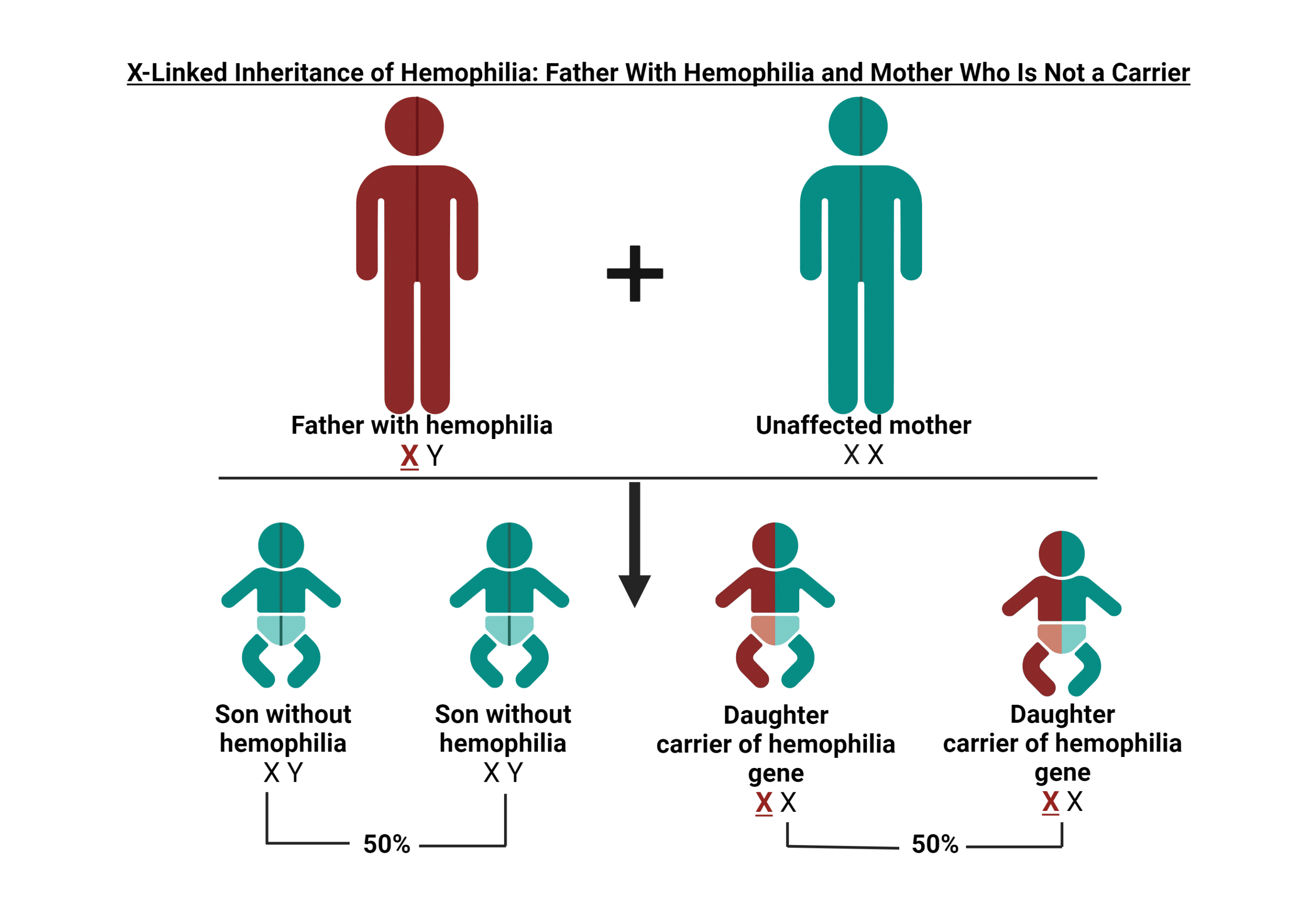
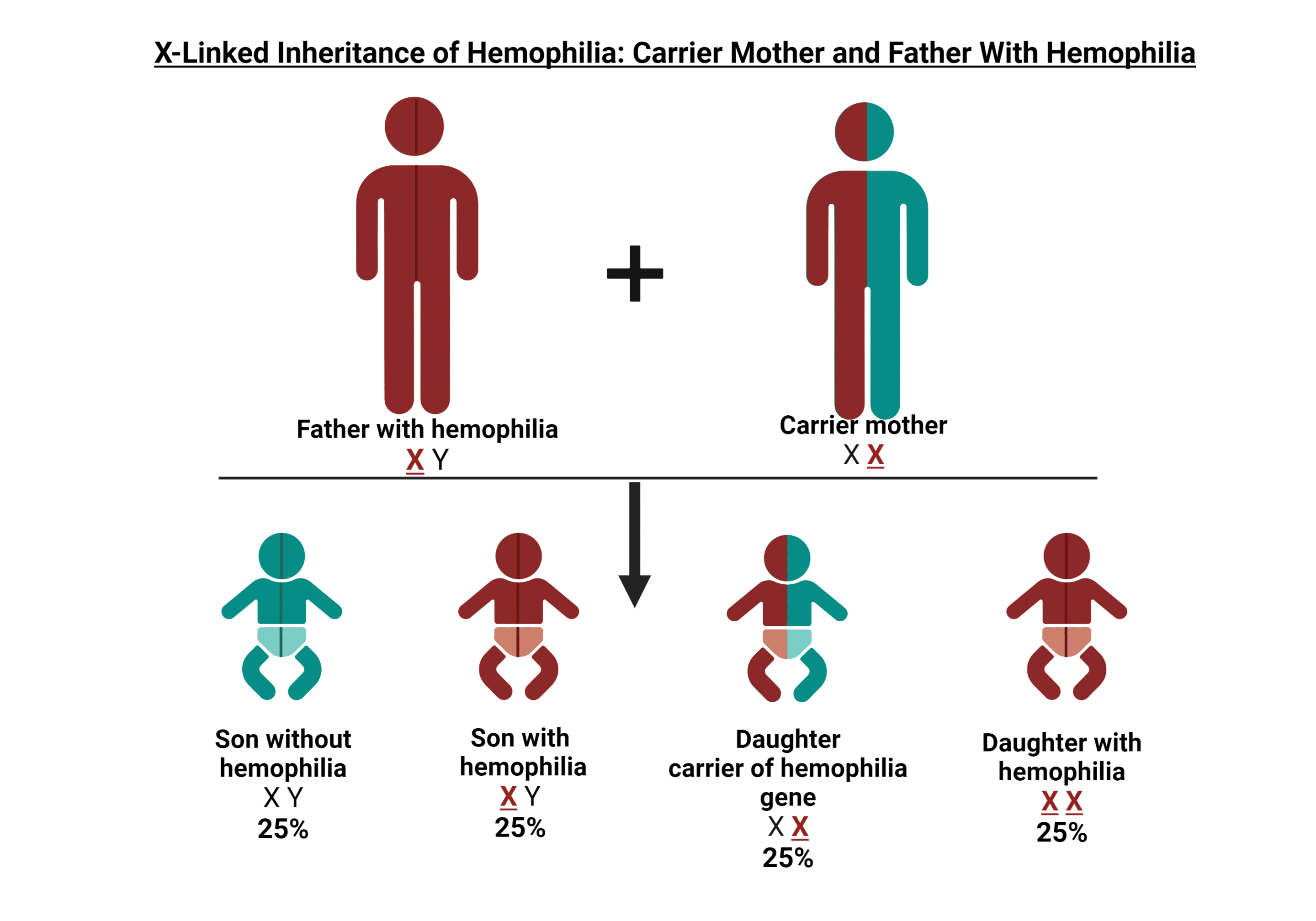

Portador de hemofilia
En aproximadamente dos tercios de los casos, hay antecedentes familiares de hemofilia. La hemofilia no solo afecta a las personas diagnosticadas directamente con la afección; Los portadores pueden experimentar síntomas y también pueden tener niveles que se consideran hemofílicos. En HOC, brindamos información valiosa y apoyo a los portadores, ayudándolos a superar los desafíos y preocupaciones únicos que conlleva ser portador de hemofilia. Nuestra atención integral va más allá de la atención médica tradicional, lo que garantiza que reciba la orientación y la atención que necesita para tomar decisiones informadas sobre su salud y bienestar y el de su familia.
¿Qué es un portador de hemofilia?
Una portadora de hemofilia es una mujer que tiene el gen que causa la deficiencia de hemofilia A (factor 8) o hemofilia B (factor 9). Este patrón de herencia genética nos ayuda a entender cómo se transmite la hemofilia en las familias.
HERENCIA GENÉTICA
Los genes de la hemofilia A y la hemofilia B se encuentran en el cromosoma X. Los hombres tienen un cromosoma X de su madre y un cromosoma Y de su padre (XY), mientras que las mujeres heredan dos cromosomas X, uno de cada padre (XX). Dado que el cromosoma Y no puede ayudar a producir el factor 8 o el factor 9 para la coagulación normal de la sangre, si un niño hereda el cromosoma X con el gen de la hemofilia de su madre, desarrollará hemofilia.
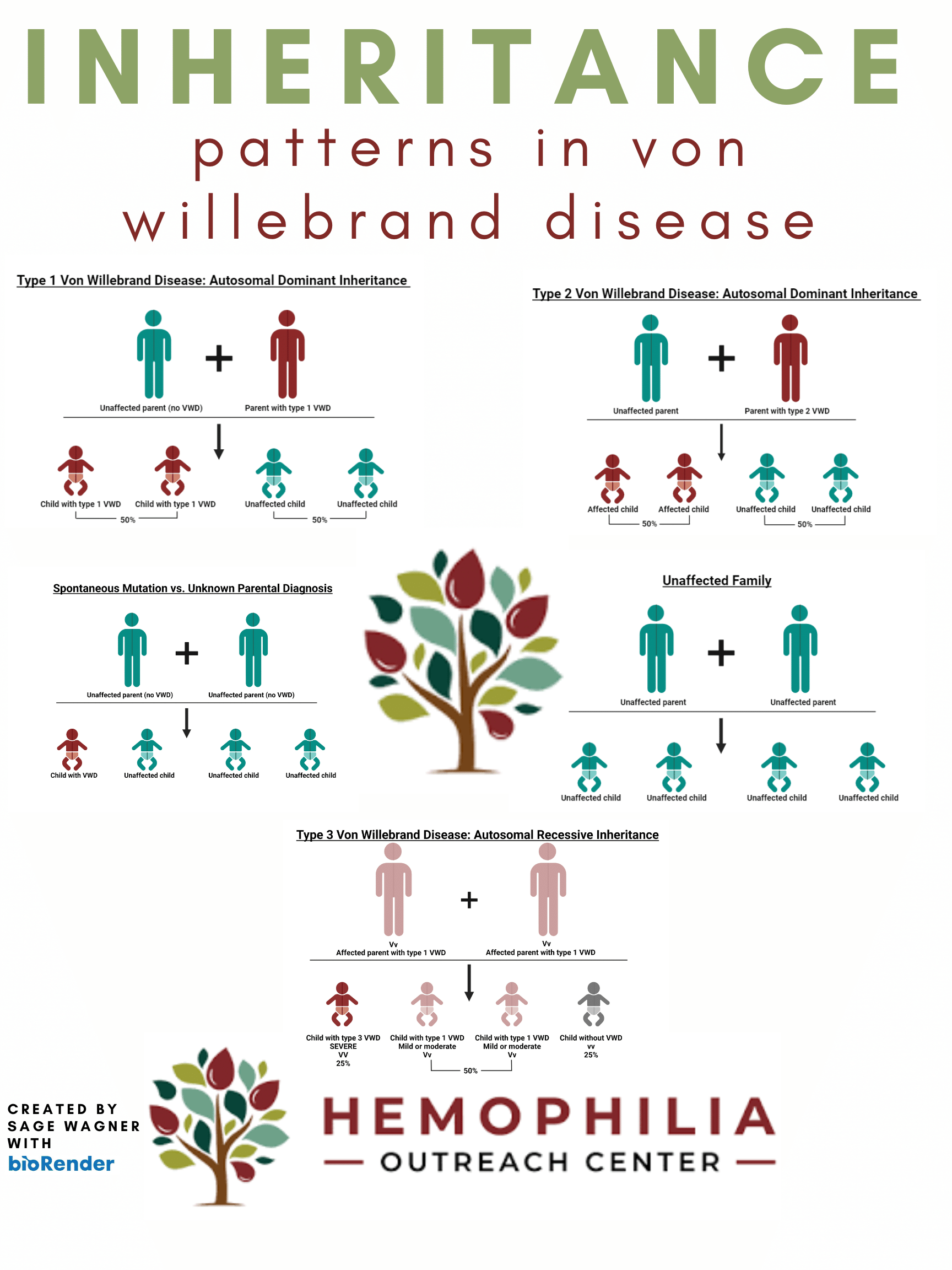
Enfermedad de von Willebrand
¿Qué es la enfermedad de Von Willebrand?
TIPOS
El factor von Willebrand (FvW) es una proteína de la sangre que desempeña un papel crucial en el proceso de coagulación sanguínea. Sirve de puente entre las plaquetas y las paredes de los vasos sanguíneos, ayudando a que las plaquetas se adhieran al vaso sanguíneo lesionado y formen coágulos para detener la hemorragia. El VWF también transporta y estabiliza un factor de coagulación llamado factor VIII, que es esencial para la coagulación de la sangre.
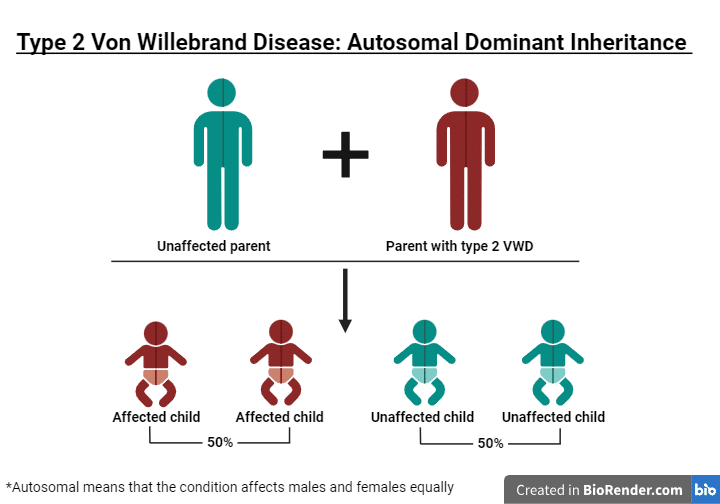
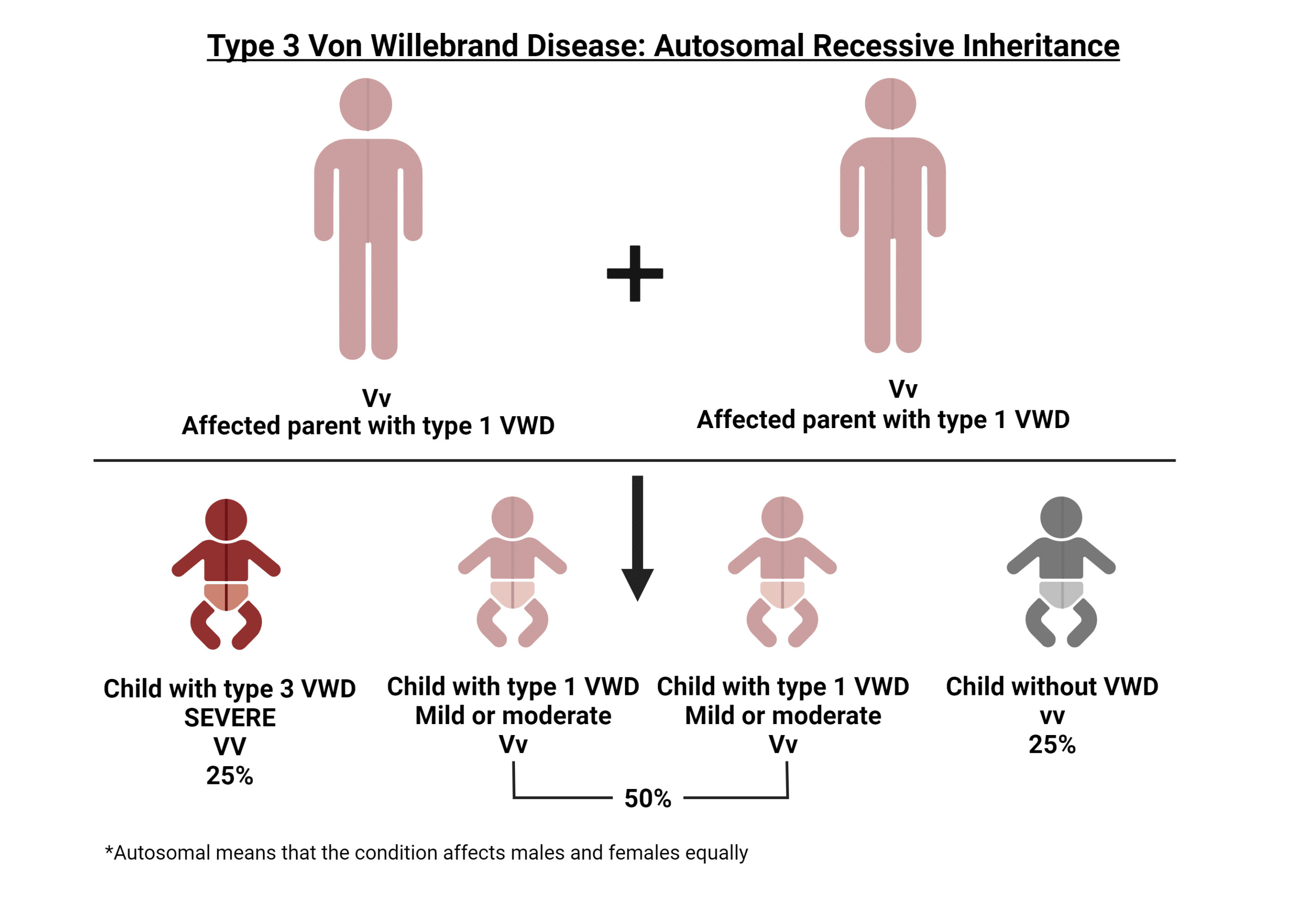
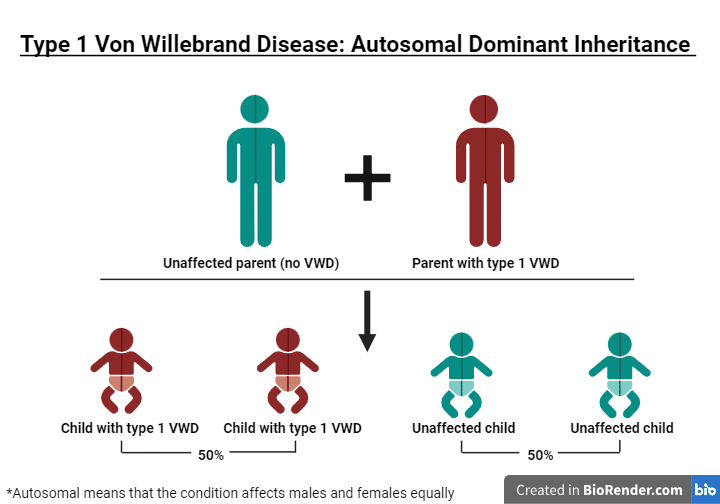
Hay tres tipos principales de enfermedad de von Willebrand. Cada tipo tiene al menos un resultado de laboratorio bajo, pero los tipos se diferencian por los multímeros. El tipo 1 tiene multímeros normales. El tipo 2 tiene multímeros anormales. El tipo 3 tiene ausencia de multímeros.
CÓMO ES DE COMÚN
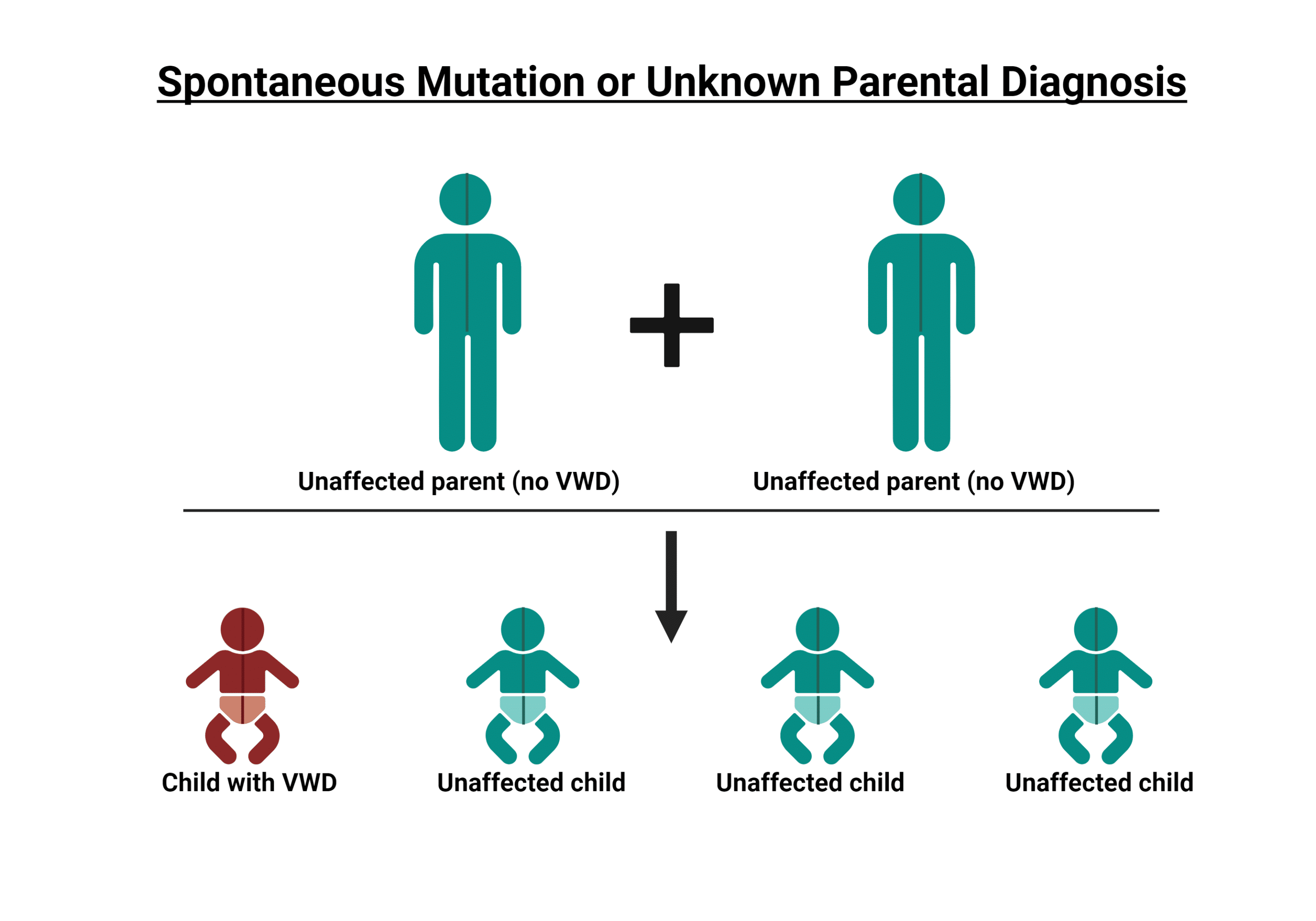
La enfermedad de von Willebrand (vWD) es mucho más frecuente y se da en aproximadamente el 1-2% de la población estadounidense. Al igual que la hemofilia, la vWD se hereda de padres a hijos. El gen anómalo de la enfermedad de von Willebrand se localiza en un cromosoma denominado autosoma, lo que significa que hombres y mujeres pueden transmitir el gen defectuoso a sus hijos. A diferencia de la hemofilia, la enfermedad de von Willebrand se da por igual en hombres y mujeres.
SEVERIDAD
Las personas con von Willebands corren el riesgo de sufrir hemorragias nasales prolongadas, hematomas con facilidad, hemorragias anormales tras intervenciones quirúrgicas, dentales o lesiones. Las mujeres son más propensas a experimentar síntomas de hemorragias abundantes o anormales durante su sangrado menstrual y después del parto. Los pacientes mayores son más propensos a sufrir hemorragias gastrointestinales crónicas.
Deficiencia plaquetaria
¿Qué es una deficiencia plaquetaria?
DEFINICIÓN
Las plaquetas son un componente crítico del proceso de coagulación de la sangre. Las plaquetas son pequeñas células en forma de disco que se crean en la médula ósea. Su función principal es ayudar a la coagulación de la sangre a través de la adhesión, la agregación, la activación y la secreción. Si hay una falla en alguna de las funciones plaquetarias habrá una mayor tendencia a sangrar.
TIPOS
Trastorno cuantitativo de las plaquetas, que es el tipo más común de deficiencia plaquetaria. Hay una disminución en el número de plaquetas que funcionan normalmente. O el cuerpo no produce lo suficiente O las plaquetas se eliminan demasiado rápido del cuerpo.
Trastorno cualitativo de las plaquetas, es el resultado de una estructura o funcionamiento anormal de la plaqueta. Las plaquetas son de mala "calidad". Algunas posibles causas incluyen la falta de proteínas o proteínas anormales en la superficie de la membrana plaquetaria. Decinecry o defectuosidad en los gránulos plaquetarios o su contenido (comúnmente conocido como trastorno del grupo de almacenamiento).
HERENCIA GENÉTICA
Las deficiencias plaquetarias pueden o no ser heredadas genéticamente, lo que significa que no es necesario tener antecedentes familiares para tener un trastorno plaquetario, pero podría transmitir el trastorno a su hijo.
Debido a la complejidad de los trastornos plaquetarios, esto suele ser difícil de diagnosticar.
SÍNTOMAS
Las personas con un trastorno plaquetario pueden experimentar moretones fáciles, hemorragias nasales, hemorragias en la boca o las encías, sangrado menstrual abundante y después del parto, y sangrado después de un trabajo dental y una cirugía.

Telangiectasia hemorrágica hereditaria (HHT)
¿Qué es la HHT?
La HHT es un trastorno en el que algunos vasos sanguíneos no se desarrollan correctamente. Una persona con HHT puede tener vasos sanguíneos malformados, concretamente vasos sanguíneos que carecen de capilares. Los capilares son vasos sanguíneos diminutos que conectan las arterias con las venas.
SÍNTOMAS
Sin los capilares -los diminutos vasos sanguíneos que conectan las arterias con las venas-, las arterias se unen directamente a las venas, lo que provoca un aumento de la presión. Este aumento de presión puede provocar hemorragias en estas zonas. La HHT se conoce comúnmente como Osler-Weber-Rendu (OWR), los nombres de varios médicos que estudiaron la HHT en el siglo XIX.
El síntoma más común en los pacientes con HHT son las hemorragias nasales que pueden o no persistir durante toda la vida de la persona. Estas hemorragias nasales ocurren en aproximadamente el 90% de los pacientes en algún momento de su vida. Las hemorragias nasales pueden ser muy poco frecuentes y pueden detenerse fácilmente u ocurrir varias veces al día.
QUÉ TAN COMÚN ES
La THH afecta aproximadamente a 1 de cada 5000 personas.


Crédito de la imagen: CureHHT
Trombastenia de Glanzmann
Trombastenia de Glanzmann
La trombastenia de Glanzmann (TG) es un trastorno hemorrágico genético causado por un mal funcionamiento de las plaquetas, que son esenciales para la coagulación de la sangre. Esta afección es el resultado de una deficiencia o disfunción del complejo glicoproteína IIb/IIIa en la superficie plaquetaria. La GT se caracteriza por tiempos de sangrado prolongados, incluso con lesiones menores, debido a la incapacidad de las plaquetas para formar coágulos adecuados. Los síntomas comunes incluyen moretones fáciles, hemorragias nasales frecuentes, encías sangrantes y, en casos graves, hemorragias internas. Se diagnostica a través de análisis de sangre especializados que evalúan la función plaquetaria y pruebas genéticas. El tratamiento de la TG implica atención preventiva, tratamiento de los episodios hemorrágicos con transfusiones de plaquetas u otros agentes y, a veces, el uso de fármacos antifibrinolíticos para reducir el riesgo de hemorragia. Vivir con TG requiere supervisión médica continua para garantizar un manejo eficaz y mantener una buena calidad de vida.

Otros trastornos hemorrágicos poco frecuentes
Hemophilia Outreach Center es su aliado dedicado en el diagnóstico y manejo de trastornos hemorrágicos raros, incluida la deficiencia del factor 7, la hemofilia adquirida, la enfermedad de von Willebrand adquirida y otros trastornos raros. Nos centramos en los resultados positivos de los pacientes y en mejorar su calidad de vida. Ya sea que necesite tratamiento clínico, apoyo para reducir los riesgos de sangrado, asesoramiento genético o una conexión con una comunidad de apoyo, HOC está aquí para empoderarlo para prosperar.
¿Qué otros trastornos hemorrágicos tratamos?
DEFICIENCIA DEL FACTOR 7
La deficiencia del factor 7 es un trastorno genético poco frecuente causado por una deficiencia o una actividad reducida del factor de coagulación 7. Este se considera el más común de los trastornos hemorrágicos raros. Se estima que ocurre en 1 de cada 300.000-500.000. Se hereda de forma autosómica, lo que significa que ambos padres deben portar el gen para transmitirlo a su hijo. Esto afecta por igual a hombres y mujeres. (cita)
Las personas con deficiencia de factor 7 corren el riesgo de sangrar después de un traumatismo, cirugía o trabajo dental. También pueden tener hemorragias internas o articulares sin una causa aparente. Cuanto más deficiente es una persona en el factor de coagulación, mayor es el riesgo de sangrado.
Las mujeres son más propensas a experimentar síntomas de sangrado abundante o anormal durante el sangrado menstrual y después del parto.
HEMOFILIA ADQUIRIDA
La hemofilia adquirida es una enfermedad autoinmune. Los anticuerpos del sistema inmunitario atacan y destruyen uno de los factores de coagulación del cuerpo (la mayoría de las veces, el factor de coagulación 8). Esto puede provocar un sangrado excesivo y puede afectar por igual a hombres y mujeres.
La hemofilia adquirida puede ser causada por otras enfermedades autoinmunes, cáncer, embarazo o ciertos medicamentos, pero otras veces no hay una causa conocida. La hemofilia adquirida es extremadamente rara, y solo afecta aproximadamente a 1 de cada 1.000.000 de personas.
CONTRAJO LA ENFERMEDAD DE VON WILLEBRAND
El sistema inmunitario también puede atacar el factor von Willebrand, lo que resulta en la enfermedad de von Willebrand adquirida.
TRASTORNO HEMORRÁGICO NO ESPECIFICADO
Las personas con trastornos hemorrágicos no especificados pueden experimentar sangrado prolongado o excesivo. Esto puede ocurrir espontáneamente, después de una lesión o durante/después de un procedimiento quirúrgico.
En estas situaciones, aunque se realizan pruebas para todos los trastornos hemorrágicos, no hay resultados de laboratorio anormales que conduzcan a un diagnóstico específico de trastorno hemorrágico.
Los pacientes suelen presentar hemorragias postquirúrgicas/dentales, hematomas por traumatismos menores, hemorragias nasales y/o ciclos menstruales abundantes.
Se desconoce la herencia de los trastornos hemorrágicos no especificados.
Es posible que se requiera tratamiento para el sangrado antes de los procedimientos, incluso sin un diagnóstico formal. Es muy importante que hable sobre sus antecedentes de sangrado anormal con todos los proveedores de atención médica y que se asegure de que HOC participe en su atención.

Cómo podemos ayudarle en su viaje por el trastorno hemorrágico
A través de nuestra atención integral y una amplia gama de servicios de salud para trastornos hemorrágicos, empoderamos a nuestros pacientes para que vivan vidas plenas y significativas. Obtenga más información sobre nuestros servicios y cómo podemos ayudarlo en su viaje hacia la hemofilia.